Provides a complete set of core features, allowing users to explore and evaluate the software.
Provides access to powerful CADD methods & algorithms, optimized for academic research and industrial projects.
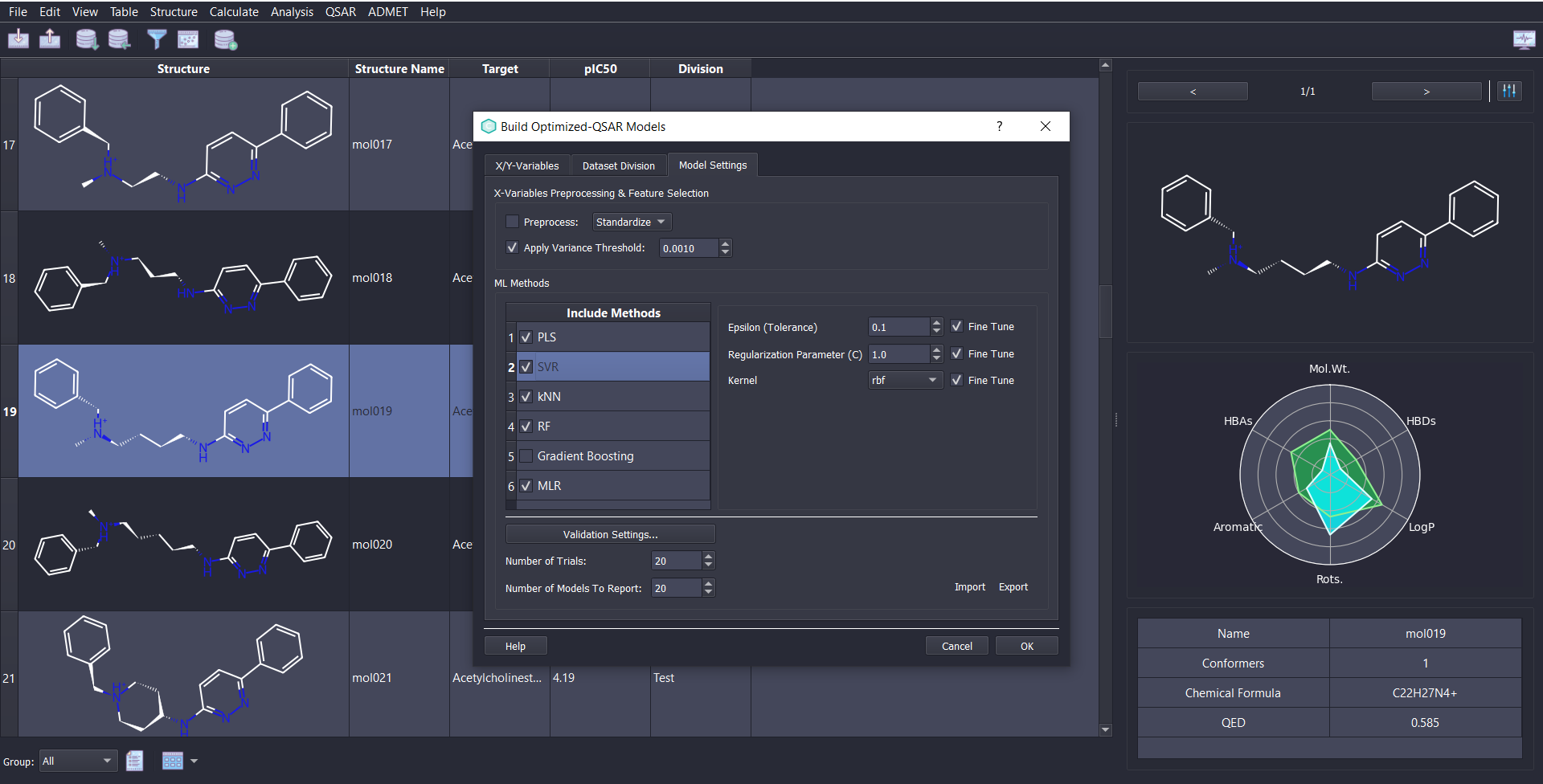
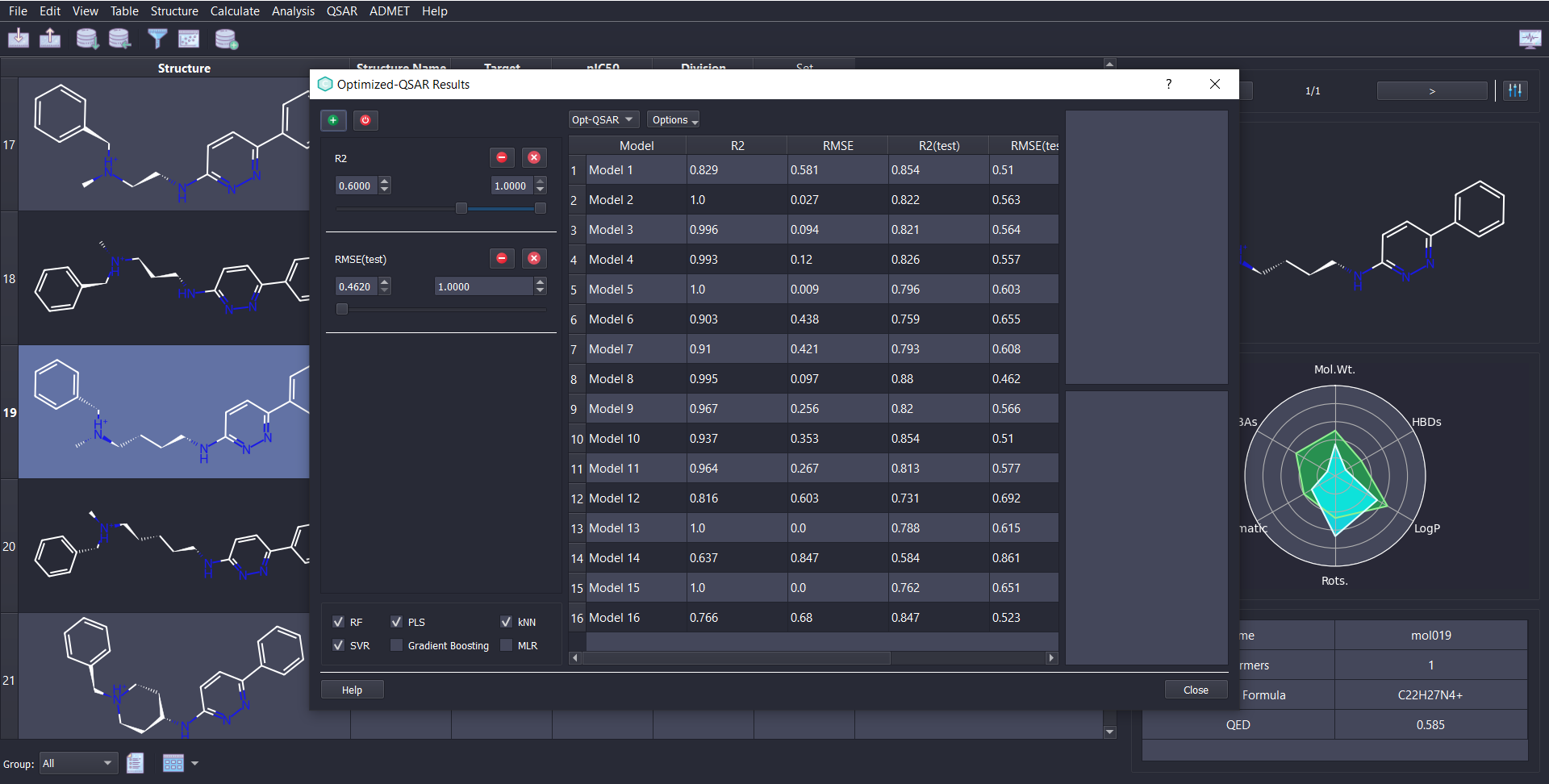
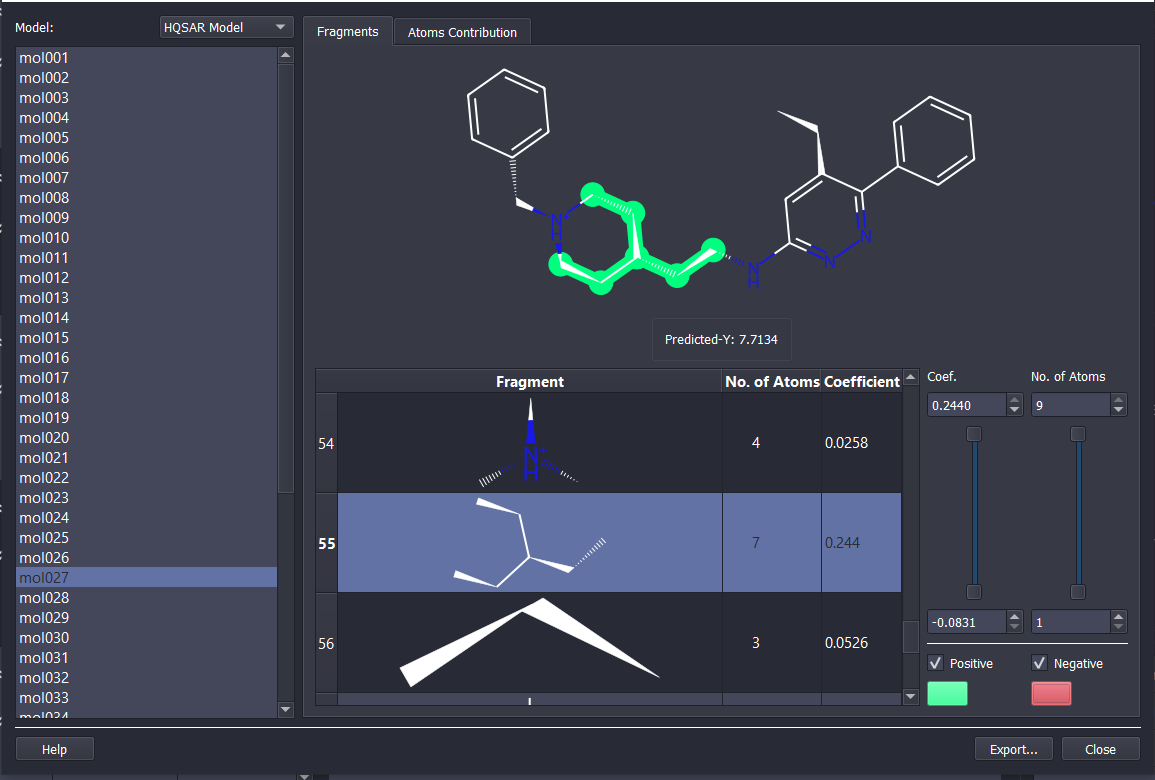
ChemMaster offers a complete and powerful toolkit for QSAR modeling, covering every step of the workflow:
Choose from diverse QSAR modules tailored to your needs—whether you require flexible control or automated modeling:
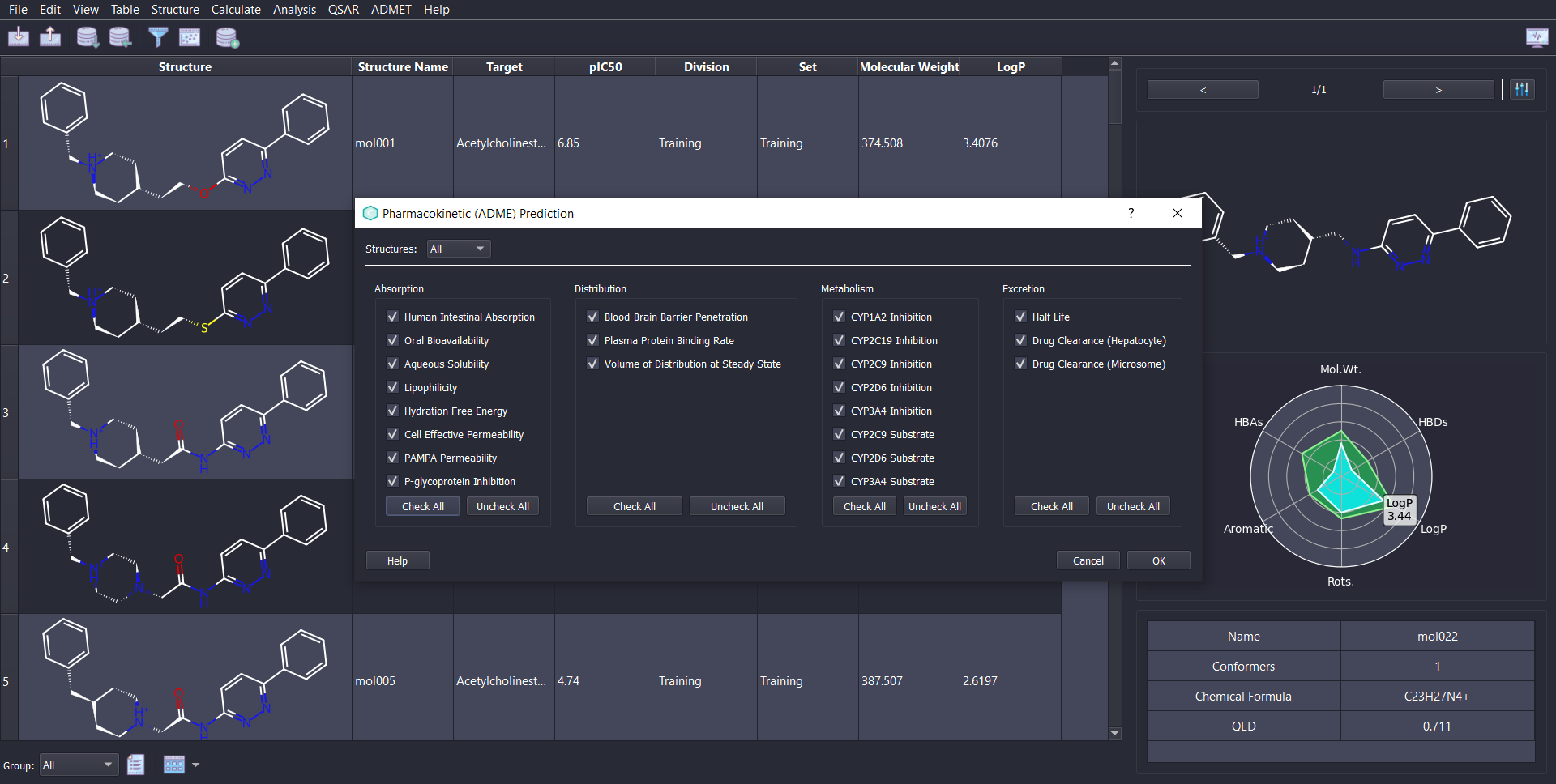
State of the art machine learning based ADMET prediction via the ADMET-AI package. ChemMaster provides an easy to use & modern user interface to utilize this powerful ADMET assessment tool and integrate it seamlessly with other types of analysis present in the software.
Generate drug-like molecules using a sophisticated genetic algorithm with control over the properties of the generated structures such as the range of molecular weight, number of rotatable bonds, etc.
Calculation of common properties used in drug design, hashed fingerprints & structural keys.
Preparation operations of structures including adding hydrogens, normalizing, removing unwanted fragments, etc. as well as management of duplicate structures.
Generation of multiple conformers using the ETKDG method.
Identification of the maximum common substructure in a set of structures with various options.
Clustering of structures using the Hierarchical clustering algorithm.
Compute similarity across various structures using molecular fingerprints.
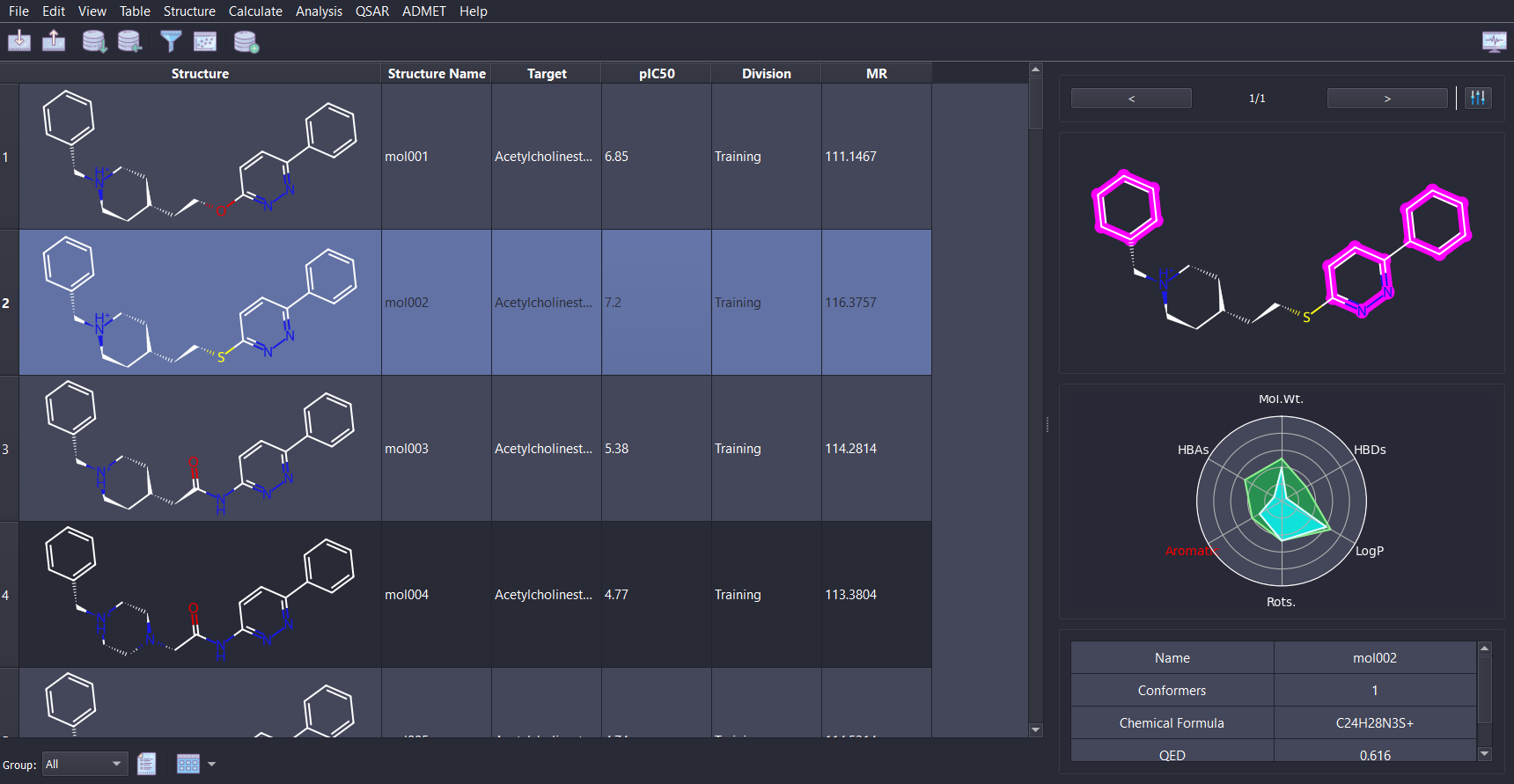
Tabulation of chemical data in a convenient spreadsheet form, allowing to manipulate the data with various processes.
SMARTS and SMILES pattern substructure search, numerical and textual filtering of the data.
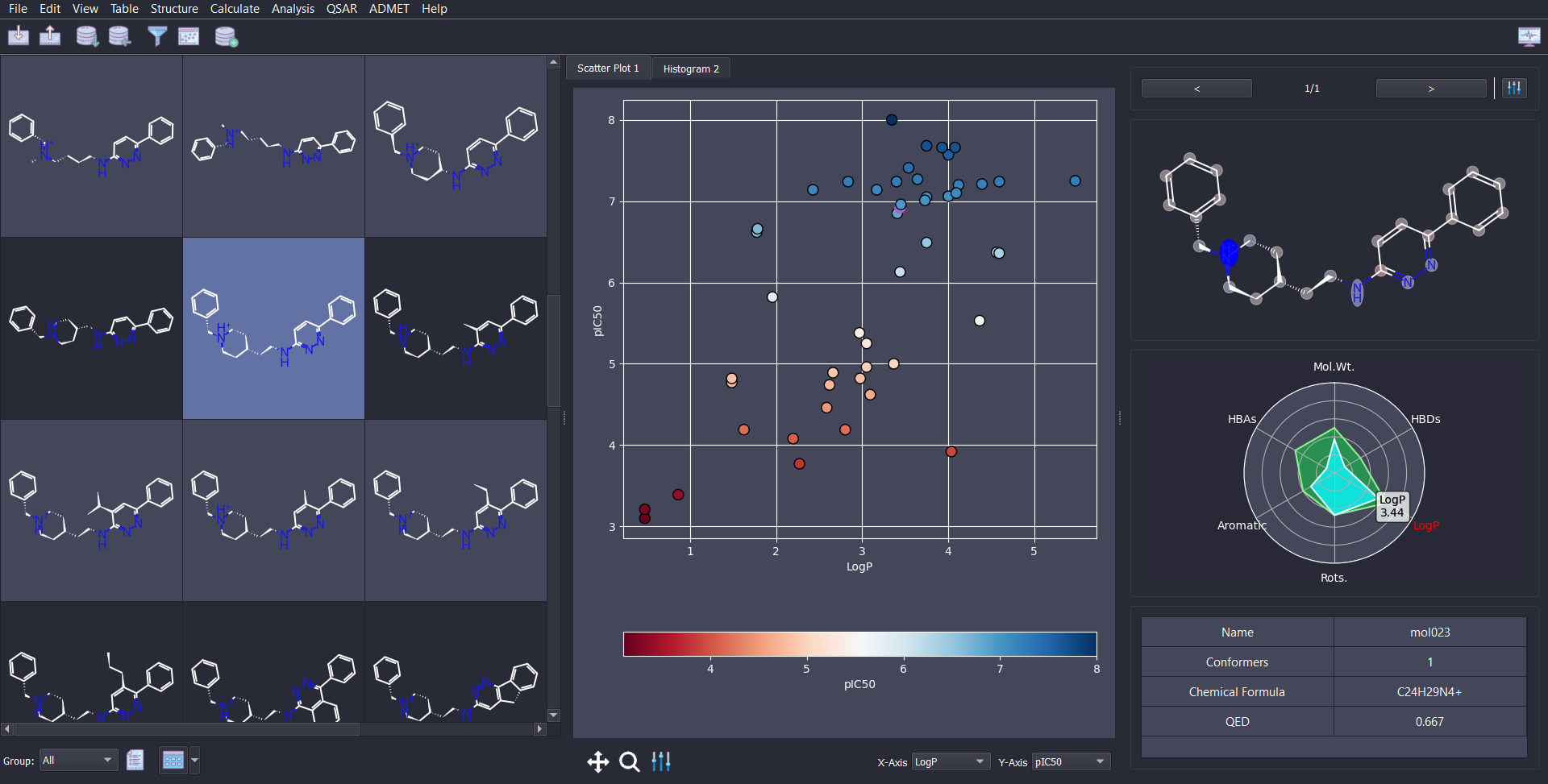
Customizable and interactive scatter and histogram plots of data.
A complete guide covering all software features with detailed parameter explanations.